Natrium – das Salz in der Suppe

2019 – Jahr des Periodensystems
Natrium ist gleichsam das Salz in der Suppe des Periodensystems.[1] Alle Menschen kennen dieses Element, wenn auch vorwiegend nicht in gediegener Form, sondern in Verbindungen. Ohne Kochsalz kein Leben, ohne Natrium-Kationen keine Reizleitung zwischen den Nervenzellen. Da wir schon lange um die Bedeutung des Salzes wissen, trachten wir seit jeher danach, es zu besitzen und führen sogar Salzkriege, z. B. 1291 um das Salzmonopol zwischen Salzburg und dem Salzkammergut.

Der Name Halit für NaCl leitet sich vom altgriechischen Namen für Salz ab, steht für Reichtum und findet sich noch heute sehr häufig wieder, wie z. B. in Städtenamen wie Bad Reichenhall, Schwäbisch Hall oder Halle (Saale). Halit ist das meistverwendete Ausgangsmaterial in der chemischen Industrie. Etwa 70% werden bergmännisch als Steinsalz abgebaut und 30% stammen aus dem Meerwasser oder Salzseen. Etwa die Hälfte der weltweit geförderten ca. 250 Mio. Tonnen werden für die Chlor-Alkali-Elektrolyse eingesetzt, um daraus Cl2, H2 und NaOH darzustellen. Die Schmelzflusselektrolyse führt zu Natrium und Chlor. Im Solvay-Prozess entsteht aus NaCl, CO2 und Ammoniak NH4Cl, NaHCO3 und letztlich Soda Na2CO3 als Flussmittel für die Glas- und Stahlindustrie, als Waschmittel, zum Wasserenthärten, als Zusatz für die Papier- und Backindustrie und vieles mehr. Das zweite Element der ersten Gruppe im Periodensystem ist gegenüber den 60 ppm für Lithium und den 15.000 ppm für Kalium mit 23.000 ppm bei weitem das häufigste Alkalimetall und das fünfthäufigste Metall überhaupt (nach Al, Fe, Ca und Mg) in der Lithosphäre. Neben den riesigen geologischen Steinsalz-Lagerstätten ist unvorstellbar viel Salz in der Hydrosphäre unseres Planeten gelöst (11.050.000 ppb Na+ und 416.000 ppb K+ gegenüber verschwindenden 180 ppb Li+). Die Weltmeere enthalten ein NaCl-Volumen von 19 Mio. km3, also 50% mehr als die Landmasse des nordamerikanischen Kontinents oberhalb des Meeresspiegels. Ein Quader Kochsalz mit der Grundfläche von einem km2 wäre 47-mal so hoch wie die Entfernung zwischen Erde und Mond.[2]
Der 29-jährige Humphry Davy entdeckte nach einem Selbststudium der Chemie 1807 das Natrium, wenige Tage nachdem er als erster Kalium rein dargestellt hatte. Er nutzte die Volta‘sche Säule (erst 1866 entwickelte Werner von Siemens den Dynamo), um beide Elemente durch Schmelzflusselektrolyse aus Natrium- oder Kaliumhydroxid darzustellen. Davor hielt man Alkalimetallsalze für elementare Reinstoffe. Bedenkt man die Reaktivität beider Metalle und die Schwierigkeiten in der damaligen Zeit, unter Luft- und Wasserausschluss zu arbeiten, so muss Davy nicht nur ein ausgesprochen fähiger, sondern auch mutiger Experimentator gewesen sein.
Eigenschaften
Wie alle Alkalimetalle ist auch Natrium ein Leichtmetall, das sich durch eine sehr geringe Dichte auszeichnet (von 0.53 g/cm3 für Lithium über 0.97 g/cm3 bis 1.90 g/cm3 für Cäsium). Natrium leitet hervorragend elektrischen Strom und Wärme. Zusammen mit dem niedrigen Schmelzpunkt von nur 98 °C (Wärmeleitfähigkeit: 140 W/mK) ist Natrium so ein sehr geeignetes Kühlmedium, z. B. in Kernreaktoren (gegenüber nur 0.56 W/mK für Wasser). Die ns1-Elektronenkonfiguration und das sehr hohe 2. Ionisationspotenzial (über 4500 kJ/mol) erklären, warum bislang keine Verbindung bekannt ist, in der Natrium mit einer höheren Oxidationsstufe als +I vorkommt. Das Metall ist weich, leicht zu formen und mit dem Messer zu schneiden. Es wird unter einer Sperrflüssigkeit aus Paraffinöl gelagert, da es mit feuchter Luft reagiert.

Natrium ist jedoch nicht so reaktiv, dass es unter Normalbedingungen mit reaktionsträgem Stickstoffgas Nitrid bildet. An Luft verbrennt das Metall zum entsprechenden Oxid. Zwei Natriumkationen passen in der Größe zum Peroxid-Anion O22–, so dass sich Na2O2 bildet. Natrium reagiert heftig mit Wasser unter Bildung von Natronlauge und Wasserstoffgas. Es bringt die Reaktionswärme auf, das Metall zu einer Kugel zu schmelzen, die dann auf einem Polster aus Wasserstoffgas auf der Oberfläche schwimmt. Wird diese Bewegung nicht unterbunden und der Abtransport der Reaktionswärme in das Wasser nicht eingeschränkt, so entzündet sich das Gas nicht. Kann sich die Kugel aus flüssigem Metall nicht frei bewegen, so wird das Knallgas explosionsartig gezündet [2]. Eine Vielzahl von YouTube-Videos oder auch ein Beitrag hier auf dieser Seite zeigt immer wieder diese gefährliche Reaktion.[3]
Ganz anders mit Ammoniak! Wird Natrium bei Temperaturen tiefer als -33°C in flüssigen Ammoniak eingebracht, bildet sich zunächst eine tiefblaue Lösung ohne Gasentwicklung.[4] Dabei wird kein Natriumamid NaNH2 gebildet, sondern das Alkalimetall-Kation und das dazugehörige Elektron als Anion in einem solvenzseparierten Ionenpaar gelöst. Der permanente Dipol NH3 ist in der Lage, mit seinem partiell negativen Stickstoffatom sowohl das Na+ zu solvatisieren, als auch mit seinen partiell positiv polarisierten Wasserstoffatomen einen Käfig zu bilden, der ein ungepaartes Elektron aufnehmen kann. Solche Lösungen aus solvatisierten Elektronen sind paramagnetisch.
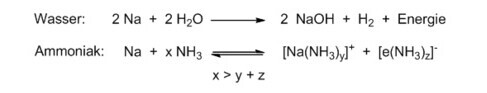
Erst bei hoher Metallkonzentration kommt es zur Ausbildung von Kontaktionenpaaren und damit zu Spinpaarung. Diese konzentrierten Lösungen sind dann goldfarben und ausgezeichnete Reduktionsmittel, um z. B. direkt aus Sauerstoff Hyperoxide oder aus Acetylen Metallacetylide herzustellen.[5]
Energiespeicher
Der uneingeschränkte König der Energiespeicher für die Elektromobilität ist gegenwärtig der Lithium-Ionen-Akku. Die Energiedichte steigt ständig bei sinkendem Preis. Aller Voraussicht nach nähert sich die Technologie jedoch einem Plateau: ein zukünftig ausgereizter Lithium-Ionen-Akku fasst voraussichtlich höchstens 30% mehr Energie pro Masse als gegenwärtig. Eine Alternative ist hier möglicherweise die Natrium-Sauerstoff-Batterie. Schon jetzt ist die Zahl der Ladezyklen höher als bei der Lithium-Sauerstoff-Batterie. Trotz viel größerer Oberfläche der Kathode aus Kohlefasern und Ether-basiertem Elektrolyten, bildet sich hier kein Natriumcarbonat[6]. In den Materialwissenschaften und der Batterieforschung spielen Schichtstrukturen und Interkalationsverbindungen eine herausgehobene Rolle. Die prominentesten Vertreter dieser Substanzklasse sind Graphitinterkalationsverbindungen durch ihren vielfältigen Einsatz als Anoden- und Kathodenmaterial in Alkalimetall-Ionen-Akkus. Selbstorganisierte Sandwich-Strukturen aus polycyclischen Carbanionen und Natriumionen, hergestellt aus verschiedensten Kohlenstoff-Allotropen, können hier als Modelle für geladene Anodenmaterialien der Zukunft dienen. Dabei sind die Schichten der Wirtsstruktur innerhalb der Ebene stark gebunden, weisen aber nur schwache Wechselwirkungen untereinander auf. Diese Schichten aus Graphit oder Übergangsmetalloxiden werden vom gewünschten Metall oder der Legierung reduziert und bilden anionische Schichten, zwischen denen sich die desolvatisierten Kationen bewegen können. Der Zusatz von Ammoniak zum Elektrolyten könnte für eine erhöhte Mobilität der Kationen sorgen. Die neuartige Schichtstruktur von [Na(NH3)4][Ind], die aus selbstorgani¬sierten abwechselnden Schichten von quadratisch-planar koordinierten Kationen und Indenylanionen besteht, könnte so ein Modellsystem sein. Sie wird im Wesentlichen durch zwei Effekte determiniert: iDe Energiedifferenz zwischen Td- und D2d-symmetrischer Form ist für Na(NH3)4+ um 25 kJ·mol-1 geringer als für Li(NH3)4+. Die niedrigere Barriere kann dabei durch NH-?-Wechselwirkungen überkompensiert werden. Die quadratisch-planare Natrium-Koordination sorgt für wenig Störung in der Schichtstruktur der Elektroden, während die NH-Π-Wasserstoffbrückenbindungen die Mobilität entlang einer Spannung erhöhen.[7]
Synthese
Neben diesen bekannten großtechnischen Prozessen spielt Natrium in der metallorganischen Synthesechemie eine zunehmend wichtige Rolle. Zwar hat es noch nicht den Stellenwert des Lithiums erreicht, holt jedoch in den letzten Jahren stark auf. Seit etwa zehn Jahren hat sich das fruchtbare Feld der heterobimetallischen Metallierungsreagenzien aus einer anfänglichen Laborkuriosität eröffnet. Mulvey et al. machten die Entdeckung, dass immer wieder cyclische Metallamidkomplexe gebildet wurden, die ein Oxid oder Peroxid im Zentrum eines Metallaheterocyclus enthielten (Abb. 3, links). Jeweils zwei einwertige Natrium- und zwei zweiwertige Erdalkalimetalle sind mit vier amidischen Stickstoffatomen verbunden. Da hier die Polaritäten gegenüber den klassischen Kronenethern invertiert sind und Anionensolvatation gegeben ist, prägten sie für diese Verbindungsklasse die Begriffe Inverse Kronenether oder Inverse Kronenkomplexe.[8]

Die Autoren konnten belegen, dass das interstitielle Sauerstoffatom aus der Etherspaltungsreaktion des Lösungsmittels stammt [9]. Eine wichtige Eigenschaft dieser Inversen Kronen ist die Fähigkeit zur Anionensolvatation. So konnten die beispiellosen Komplexe [Na4Mg2(TMP)6(C6H3Me)] und [Na4Mg2(TMP)6(C6H4)] (TMP = Tetramethylpiperidin, Abb. 3 Mitte und rechts) erhalten werden. Der erste enthält ein zweifach deprotoniertes Toluol-Molekül als Dianion im Zentrum der Inverskrone und der andere gar ein zweifach deprotoniertes Benzolmolekül. Trotz des geringeren pKs-Wertes der Kernwasserstoffatome verglichen mit der Methylgruppe (38 gegenüber 40), wird durch das Gemisch BuNa, Bu2Mg und TMP(H) der Ring in der ortho- und meta-Position deprotoniert. Sogar der König der Aromaten, das ansonsten inerte Benzol (pKs-Wert = 43), wird zweifach deprotoniert. Diese Reaktivität ist weit außerhalb der Reichweite für Organolithiumverbindungen. Keine Komponente allein könnte die Deprotonierung des aromatischen Kerns von Arenen bewerkstelligen. Die doppelte Π- und Π-Stabilisierung eines deprotonierten aromatischen Produkts durch ein Alkali- und Nichtalkalimetall ist das typische Markenzeichen der Alkali Metal Mediated Metallation (AMMM, der Alkali Metall vermittelten Metallierung).[10] In nicht-synergetischen Systemen sind Organonatrium-Verbindungen um Größenordnungen reaktiver als Diorganomagnesium-Verbindungen, aber in diesen Cokomplexen führt das weniger reaktive Magnesium die Deprotonierung aus und bildet die Mg-C-Π-Bindung in der Ringebene aus, während das Natrium, notwendig für die Deprotonierung, nur die Π-Beobachterrolle einnimmt. Setzt man zwei Äquivalente Metallierungsreagenz ein, so wird Benzol in der sterisch bevorzugten 1,4-Position dimagnesiert, jetzt mit beiden Natriumatomen Π-gebunden.
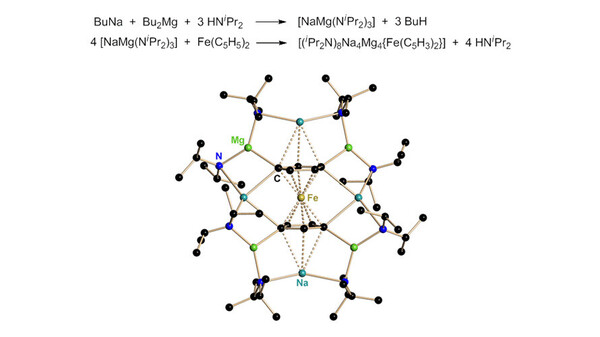
Einer der größten Erfolge dieser gemischtmetallischen Na/Mg Amide ist jedoch sicher die Tetradeprotonierung des Ferrocens. [11] Mit Organolithiumverbindungen und geeigneten Donorbasen kann man allenfalls die 1,1‘-Dimetallierung des Cp2Fe (Cp = Cyclopentadienyl, C5H5-) erreichen. Damit ist die Derivatisierung dieser wichtigen Verbindungsklasse deutlich limitiert. Beide Cyclopentadienylringe von Metallocenen ließen sich bislang nur monosubstituieren. Mit [NaMg(NR2)3] geling jedoch die 1,1‘,3,3‘-Tetrametallierung vieler Metallocene (Abb. 4). In der Struktur des [(iPr2N)8Na4Mg4{Fe(C5H3)2}] ist das zentrale [Fe(C5H3)2]4-Tetraanion von einem Torus aus alternierend acht Metallkationen und acht Π-verbrückenden Amiden umgeben. Die deprotonierten Kohlenstoffatome verbrücken dabei jeweils ein Natrium- und ein Magnesium-Kation. Zwei weitere Natrium-Kationen sind an die beiden Cp-Ringe η3-gebunden.
Literatur
Lehrvideos zum Natrium unter http://www.uni-goettingen.de/de/lehrvideos/550700.html
D. Stalke, Praxis der Naturwissenschaften – Chemie in der Schule 2014, 63, 5-13.
Lehrbücher wie a) A. F. Holleman, N. Wiberg, Lehrbuch der Anorganischen Chemie, 102. Auflage, de Gruyter, Berlin, 2007, S. 1259-1299; b) N. N. Greenwood, A. Earnshaw, Chemistry of the Elements, 2. Auflage, Elsevier, Oxford, 1997, S. 68-106; c) C. E
Lehrmaterial-DVDs unter www.lerngut.com/produkte/unterrichtsmaterial-s-block-elemente-dvd-/
Schon Davy beobachtete das Phänomen 1807, aber W. Weyl publizierte es dann in Ann. Phys. 1864, 197, 601-612.
a) P. Hartmann, C. L. Bender, M. Vracar, A. K. Dürr, A. Garsuch, J. Janek, P. Adelhelm, Nat. Mat. 2013, 12, 228-232; b) R. v. Noorden, Nature, 2014, 507, 26-28.
R. Michel, T. Nack, R. Neufeld, J. M. Dieterich, R. A. Mata, D. Stalke, Angew. Chem. 2013, 125, 762-766; Angew. Chem. Int. Ed. 2013, 52, 734-738.
R. E. Mulvey, Chem. Commun. 2001, 1049-1056.
R. E. Mulvey, V. L. Blair, W. Clegg, A. R. Kennedy, L. Russo, Nat. Chem. 2010, 2, 588-591.
R. E. Mulvey, Acc. Chem. Res. 2008, 42, 743-755.
W. Clegg, K. W. Henderson, A. R. Kennedy, R. E. Mulvey, C. T. O’Hara, R. B. Rowlings, D. M. Tooke, Angew. Chem. 2001, 113, 4020–4023; Angew. Chem. Int. Ed. 2001, 40, 3902–3905.

Prof. Dr. Dietmar Stalke
Institut für Anorganische Chemie Universität Göttingen
Das Periodensystem ist ein faszinierendes Ordnungssystem, das die Natur den Elementen gegeben hat. Vor 150 Jahren wurde dieses System erstmals von Wissenschaftlern erkannt. Die Generalversammlung der Vereinten Nationen und die UNESCO haben das Jahr 2019 daher zum International Year of the Periodic Table of Chemical Elements, dem Internationalen Jahr des Periodensystems ausgerufen. Die Elemente des Periodensystems werden in loser Folge vorgestellt.
Kommentare
Keine Kommentare gefunden!