Supramolekulare Polymere

100 Jahre Makromolekulare Chemie
Vor hundert Jahren bewies Hermann Staudinger das Vorliegen kovalenter Bindungen für zahlreiche natürliche und synthetische Polymere. Dies führte zu einem Paradigmenwechsel, welcher den Gegenentwurf einer supramolekularen Polymerisation zunächst wenig aussichtsreich erscheinen ließ. So standen organische wie makromolekulare Chemiker gerade in Deutschland der von Jean-Marie Lehn in den 1990er Jahren sehr breit formulierten Disziplin der supramolekularen Chemie1 eher kritisch gegenüber. Sie sahen die Chemie der nicht-kovalenten Assoziate („self-assembly“) skeptisch. Wozu sollten reversible nicht-kovalente Bindungen nützlich sein? Schließlich waren die mit ihnen aufgebauten Systeme doch intrinsisch instabil, so dass erhöhte Temperaturen oder Änderungen des Lösungsmittels zur Dissoziation in Monomere führen.

Abbildung 1: Strukturen von kovalenten und supramolekularen Polymeren. © Frank Würthner
Polymerchemie mit nicht-kovalenten Bindungen
Unter den zahlreichen unter dem Begriff „self-assembly“ bearbeiteten supramolekularen Systemen, zu denen molekulare Kapseln oder Mizellen (nulldimensional), Monolagen auf Oberflächen (zweidimensional) sowie Flüssigkristalle (dreidimensional) zählen, nehmen die supramolekularen Polymere (eindimensional) eine besondere Rolle ein: Sie stehen den typischen kovalenten Polymeren strukturell am nächsten. Allerdings mit dem Unterschied, dass die Bindungen zwischen den einzelnen monomeren Bausteinen nicht-kovalenter Natur sind. Solche supramolekulare Polymere stellen somit exakt den Gegenentwurf zu den von Staudinger erstmals zweifelsfrei bewiesenen Makromolekülen dar (Abbildung 1).
Die Entwicklung supramolekularer Polymere
Anhand von Abbildung 2 sollen im Folgenden wichtige Meilensteine in der Entwicklung supramolekularer Polymere beschrieben werden. Als frühes Beispiel können hier die Arbeiten von Günter Scheibe angesehen werden. Er berichtete in seinem Vortrag vor der Münchner Chemischen Gesellschaft 1936 erstmals von der reversiblen Gelatinierung von Pseudoisocyanin-Farbstoffen und schrieb die dabei beobachteten Änderungen in den Absorptionsbanden „polymerisierten Farbstoffmolekülen“ zu.2 Diese J-Aggregate sowie eine größere Zahl anderer Farbstoffaggregate waren insbesondere in wässrigen Medien somit seit langer Zeit bekannt.

Abbildung 2: Meilensteine auf dem Gebiet der supramolekularen Polymere. © Frank Würthner
Bedingt durch den starken Einfluss des Wassers („hydrophober Effekt“) auf die Aggregatstruktur entsprachen die J-Aggregate aber nicht dem supramolekularen Denkansatz. Nach diesem sollte die Strukturbildung präzise mittels nicht-kovalenter Wechselwirkungen realisiert werden. Hierfür machte Jean-Marie Lehn 1990 einen Vorschlag unter Einsatz komplementärer Wasserstoffbrückenbindungen. Die Schwäche der von Lehn gewählten Dreifachwasserstoffbrückenbindungen machte die Herstellung supramolekularer Polymerketten in verdünnten Lösungen jedoch aussichtslos. Die geplanten Strukturen konnten lediglich in den lösungsmittelfreien flüssigkristallinen Phasen als strukturgebendes Motiv nachgewiesen werden.1 Insofern konnte hier ähnlich wie bei den bereits zuvor von Helmut Ringsdorf und anderen untersuchten diskotischen Flüssigkristallen nur bedingt von supramolekularen Polymeren gesprochen werden.3 Das Konzept erwies sich aber als tragfähig, sollten doch in den folgenden Jahren in rascher Abfolge supramolekulare Polymere auf Basis verschiedener nicht-kovalenter Wechselwirkungen realisiert werden.4 Ausschlaggebend für den Erfolg war die Entwicklung thermodynamisch ausreichend starker Bindungseinheiten für die jeweils zum Einsatz kommenden Lösungsmittel. Durch diese ließen sich in Lösung ausreichende Polymerisationsgrade für das Auftreten polymertypischer Eigenschaften wie eine starke Viskositätszunahme erreichen. Beispielhaft seien hier die auf Vierfach-Wasserstoffbrückenbindungen basierenden selbstkomplementären Ureidopyrimidon-Systeme von Bert Meijer sowie die Koordinationspolymere von Matthias Rehahn genannt. Diese beiden Beispiele zeigen auch schön die Analogie zu Kondensationspolymeren, welche wie im Falle von Perlon und Nylon entweder aus einem oder aus zwei Komponenten aufgebaut werden.
Aktuelle Trends – Kontrollierte supramolekulare Polymerisationen
In diesen frühen Arbeiten stand die thermodynamische Kontrolle der supramolekularen Polymerisation im Vordergrund. Erst die Entwicklung stärkerer supramolekularer Bindungseinheiten, welche sich insbesondere aus der Kombination von Wasserstoffbrückenbindungen und der Wechselwirkung ausgedehnter aromatischer π-Systeme ergeben, eröffnete neue Perspektiven für eine kinetische Polymerisationskontrolle (Abbildung 3).5 Unter optimierten Bedingungen konnten so supramolekulare Polymerisationen 2014/15 erstmals „lebend“ realisiert werden, indem neue Monomere sich sehr viel schneller an die Kettenenden eines bereits bestehenden supramolekularen Polymers anlagern als durch Selbstassoziation neue Ketten zu bilden. Das supramolekulare Design der für lebende supramolekulare Polymerisationen infrage kommenden Monomere ist nicht trivial. Es wird aber gegenwärtig von den ersten erfolgreichen Synthesen von supramolekularen Block-Copolymeren angespornt.5
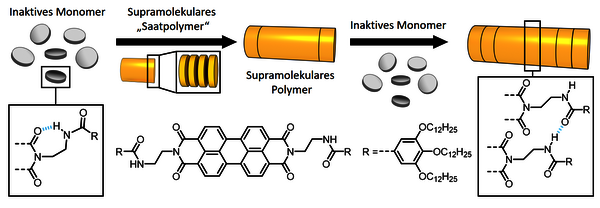
Abbildung 3: Beispiel für eine lebende supramolekulare Polymerisation. © Frank Würthner
Auch wenn hier nur ein Bruchteil der bekannten supramolekularen Polymere vorgestellt werden konnte, so lassen diese Beispiele bereits erahnen, was für ein großer Substanzraum unter dem Begriff der supramolekulare Polymere zusammengefasst wird. Aufgrund der gegenüber kovalenten Bindungen schwächeren Verknüpfung der Bausteine und deren starker Abhängigkeit vom Medium, stellen supramolekulare Polymere im allgemeinen keine Konkurrenten zu konventionellen Polymeren für die für diese etablierten Anwendungsfelder dar. Die größten Anwendungsperspektiven für supramolekulare Polymere dürften wohl im Bereich der Funktionspolymere liegen. Sie beinhalten photovoltaische Anwendungen von über π–π-Kontakte miteinander verknüpften Farbstoffen („Supramolekulare Elektronik“), elektrochrome Koordinationspolymere sowie auf supramolekularen Hydrogelen basierende Materialien für Anwendungen in der Medizin. Bemerkenswert ist die Tatsache, dass bereits die ersten supramolekularen Polymere von Scheibe zu Marktprodukten geführt haben. Denn J-Aggregate erwiesen sich als ideal geeignet für die spektrale Photosensibilisierung von Silberhalogenid in der Farbphotographie. Die strukturelle und funktionale Verwandtschaft der Scheibe’schen Cyaninfarbstoffaggregate mit denen der Chlorophyllaggregate in natürlichen Lichtsammelsystemen der Photosynthese wurde dagegen erst sehr viel später erkannt.
Autor: Prof. Dr. Frank Würthner,
Universität Würzburg, Center for Nanosystems Chemistry
Redaktionelle Bearbeitung: Maren Mielck, GDCh
Literatur:
1 J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, Wiley-VCH 1995
2 G. Scheibe, L., Kandler, H. Ecker, Naturwissenschaften 1937, 25, 75: Polymerisation und polymere Adsorption als Ursache neuartiger Absorptionsbanden von organischen Farbstoffen.
3 H. Ringsdorf, B. Schlarb, J. Venzmer, Angew. Chem. 1988, 100, 117-162: Molekulare Architektur und Funktion von polymeren orientierten Systemen – Modelle für das Studium von Organisation, Oberflächenerkennung und Dynamik bei Biomembranen
4 L. Brunsveld, B. J. B. Folmer, E. W. Meijer, R. P. Sijbesma, Chem. Rev. 2001, 101, 4071−4098. Supramolecular Polymers.
5 M. Wehner, F. Würthner, Nat. Rev. Chem. 2020, 4, 38-53: Supramolecular polymerization through kinetic pathway control and living chain growth.
Kommentare
Keine Kommentare gefunden!