Molekulare Einblicke in Polymerwerkstoffe

100 Jahre Makromolekulare Chemie
Polymere werden überwiegend als Werkstoffe eingesetzt, deren einzigartige mechanische Eigenschaften im Vordergrund stehen. Gerade im Bereich der Hochleistungspolymere spielen strukturelle und morphologische Eigenschaften die zentrale Rolle. Wichtige Aspekte sind z.B. eine makroskopische Orientierung der Kettenmoleküle, eine Strukturierung in Bereiche unterschiedlicher Mobilität oder molekularer Ordnung auf der Nanometerskala oder eine Kombination aus diesen. Eine nanoskalige Strukturierung ist vielen relevanten Werkstoffen aufgrund ihrer chemischen Struktur molekular „eingeprägt“ – dies trifft prominent für alle teilkristallinen Polymere sowie Blockcopolymere zu. Als Beispiel sei die Schlagzähmodifikation von glasartig-sprödem Polystyrol (PS) durch Pfropfung mit kautschukartigem Polybutadien (PB) erwähnt. Dabei liegt letzteres phasensepariert vor und bildet nanoskalige Einschlüsse, welche die Rissbildung eindämmen.

Abb. 1: Atomkraft- (a) und Transmissionselektronenmikroskopische Aufnahmen (b) einer schlagzähen Mischung von Polystrol und einem PS-PB Blockcopolymer [Thomann et al., Macromolecules 2009, 42, 5684]. Bild: Copyright
Es wird damit deutlich, dass ein rationales Verständnis von Werkstofffunktion nur durch Charakterisierungsmethoden zu erreichen ist, die Strukturen im Nanometerbereich auflösen und damit quantifizierbar machen. Gleichzeitg müssen sie in der Lage sein, über die molekulare Mobilität in den jeweiligen Bereichen Auskunft zu geben. Gerne wird hier mikroskopischen Methoden der Vorzug gegeben (s. Abb. 1), die zwar einen scheinbar objektiven Eindruck vermitteln, aber im erforderlichen Auflösungsbereich durchaus aufwendig sind, und vor allem kein globales, statistisch relevantes Bild liefern. In diesem Beitrag möchten wir daher einerseits die Vorteile der Protonen(1H)-Niederfeld-Kernresonanspektroskopie (low-field nuclear magnetic resonance, LF-NMR) herausstellen und die komplementäre Verwendung von Streumethoden, vor allem der Röntgenkleinwinkelstreuung (small-angle X-ray scattering, SAXS), illustrieren.
LF-NMR
Die LF-NMR liefert mit geringem Aufwand ein qualitatives Bild der Mobilität der morphologischen Komponenten und ihren quantitativ bestimmbaren Anteil in einer Probe. Abb. 2 zeigt am Beispiel des Materials aus Abb. 1 den Zusammenhang zwischen einem hochaufgelösten 1H-Festkörper-NMR-Spektrum und einem LF-NMR-Signal in der Zeitdomäne. Unter den verwendeten Messbedingungen (v.a. bzgl. Magnetfeldstärke und Geschwindigkeit des „magic-angle spinning“, MAS) sind nur die Signale des Weichanteils im Blockcopolymer (v.a. PB) spektral gut aufgelöst, während der Hartanteil (v.a. PS) nur als sehr breites Untergrundsignal zu sehen ist. Prinzipiell kann das Signalverhältnis (=Protonenverhältnis) durch Integration bestimmt werden. Dieser Weg ist aber technisch sehr aufwendig und zudem aufgrund technischer Gegebenheiten eines Hochfeldgerätes (v.a. Basislinienprobleme) ungenau.
An dieser Stelle spielt die LF-NMR in der Zeitdomäne ihren Vorteil aus. Man verzichtet auf eine spektrale Auflösung und konzentriert sich ganz auf den großen Unterschied in den T2-Relaxationszeiten der Komponenten, die prinzipiell die Breite der Spektralpeaks bestimmen, aber in der Zeitdomäne durch Kurvenanpassung viel genauer ermittelt werden können. Der breite Spektralpeak des PS entspricht der Komponente mit sehr kurzem T2, also dem Anfangsabfall. Die Amplitude desselben nimmt wie in Abb. 2b zu sehen ist, mit steigender Temperatur signifikant ab. Dies wurde mit einer variablen Mischbarkeit der beiden Polymerkomponenten an der Grenzfläche zwischen den Phasen interpretiert. Mit anderen Worten weist die Temperaturabhängigkeit der glasartigen und weichen (kautschukartigen) Anteile auf die Existenz einer gemischten Grenzschicht mit modifizierter Glasübergangstemperatur hin.
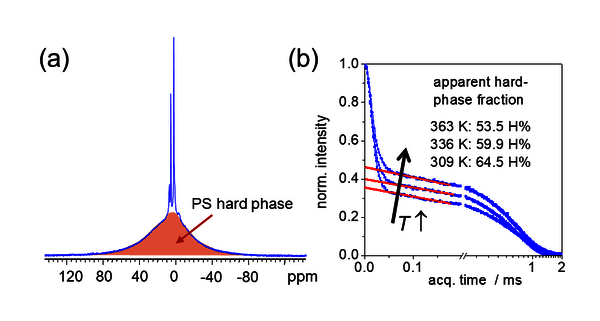
Abb. 2: 1H-Hochfeld- (a) und Niederfeld-NMR-Daten (b) der Probe aus Abb. 1. Das sog. „magic-angle-spinning“ (MAS) Spektrum in (a) zeigt das glasartige PS als breites Signal. In der Zeitdomäne am Niederfeld (b) ist der Hartanteil leicht als Signalanteil der Komponente mit kurzer T2-Relaxationszeit zu erkennen und durch einfaches Ablesen abzuschätzen.
SAXS
Während die NMR sensitiv für unterschiedliche molekulare Mobilität ist, wird das Signal in SAXS durch nanoskalige Bereiche unterschiedlicher Dichte verursacht, die in vielen Fällen den in der NMR detektierten Bereichen entsprechen. Im Gegensatz zur NMR erlaubt die Röntgenstreuung nun die Bestimmung von Strukturgrößen, wobei periodische Strukturen sich als im Peaks in der Streuintensität zeigen, während ungeordnete Strukturen i. A. zu einem diffusen Streusignal führen, das mit zunehmenden Streuvektor s = 2 sin θ / λ abnimmt. Hier ist θ der halbe Streuwinkel. Bei in Abb. 3 gezeigten Messungen an Blockcopolymeren lässt sich aus der Position des Peaks die typische Länge der näherungsweise periodisch angeordneten Bereiche aus den beiden Komponenten des Blockcopolymeren bestimmen, L = 1/smax. Aus der Tatsache, dass sich L mit Zugabe eines 60 % Mischungsanteils von reinem PS nicht vergrößert, lässt sich sofort schließen, dass sich eine inhomogene Struktur einstellt mit größeren Bereichen, in denen das PS-PB Blockcopolymer und PS getrennt vorliegen (siehe auch Abb. 1).
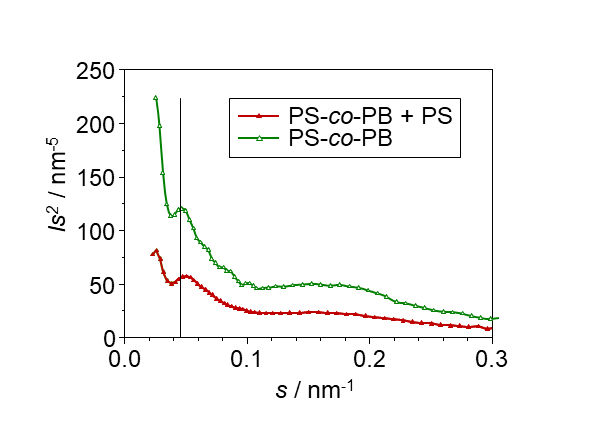
Abb. 3: SAXS-Daten der Polymermischung aus Abb.1 im Vergleich mit dem reinen PS-PB-Copolymer, welche eine direkte Bestimmung der Periodizität erlauben.
In anderen Systemen, in denen regelmäßigere Strukturen vorliegen, lässt sich das Signal der Röntgenkleinwinkelstreuung auch in viel größerem Masse quantitativ auswerten. Dies ist insbesondere bei teilkristallinen Polymeren sehr gut möglich. Polymere bilden bei der Kristallisation aus der Schmelze typischerweise eine nanoskalige, näherungsweise periodische Struktur aus, bestehend aus dünnen Schichtkristallen getrennt durch ungeordnete amorphe Bereiche. Diese mikroskopische Struktur bestimmt wesentlich die mechanischen Eigenschaften dieser Materialien. Eine Beispielmessung ist in Abb. 4 gezeigt, die das gemessene Streusignal im Vergleich mit einer Modellrechnung zeigt. Durch Anpassung der Modellparameter lassen sich die Strukturgrößen, also die Dicke der kristallinen und amorphen Bereiche, bestimmen. Aktuellere Verbesserungen ermöglichen sogar einen Zugang zu den Verteilungsbreiten dieser Strukturgrößen, wodurch auch Informationen über Inhomogenitäten gewonnen werden können.

Abb. 4: SAXS-Daten des teilkristallinen Polymers Poly(oxymethylen), Delrin® 500P, isotherm kristallisiert bei 165°C. angepasst mit einer Modellfunktion basierend auf einem Stapelmodell aus alternierenden kristallinen und amorphen Schichten. Die Anpassung der Modellfunktion erlaubt die Bestimmung der Strukturparameter und sogar ihrer Verteilungsbreiten.
Zusammenfassung
Mit den gezeigten Beispielen möchten wir vor allem die hohe Komplementarität der beiden vorgestellten Methoden herausstellen. Man erhält mit NMR und SAXS aus völlig unterschiedlichen Blickwinkeln (Mobilität bzw. Elektronendichtekontrast) in vielen Fällen entweder quantitative vergleichbare oder sich ergänzende Informationen. So konnten wir in vielen Arbeiten bestätigen, dass der Kristallinitätsgrad teilkristalliner Polymere, den man mittels LF-NMR aus dem hart/weich-Signalverhältnis gewinnt, in der Regel mit der SAXS-basierten „linearen Kristallinität“, also dem Verhältnis von Lamellendicke zu Langperiode, übereinstimmt. Charakteristische Abweichungen, wie im Beispiel des PS-PB-Blockcopolymers in Abb. 1–3 gezeigt, lassen wiederum Rückschlüsse auf Besonderheiten an den Grenzflächen solcher mehrphasigen Materialien zu. Damit lässt sich schließen, dass eine komplementäre Verwendung beider Methoden immer zusätzliche wertvolle Informationen liefert.
Autoren: Prof. Dr. Kay Saalwächter, Prof. Dr. Thomas Thurn-Albrecht,
Martin-Luther-Universität Halle-Wittenberg
Redaktionelle Bearbeitung: Maren Mielck (GDCh)
Kommentare
Keine Kommentare gefunden!